Hema Manvi Koneru, MBBS, Rajiv Gandhi Institute of Medical Sciences, Telangana, India
Divyasri Koneru, MBBS, Dr.Pinnamaneni Siddartha Institute of Medical Sciences and Research Foundation, Andhra Pradesh, India.
Amanpreet Kaur, MBBS, Government Medical College, Patiala, Punjab, India.
Amin H. Karim MD Baylor College of Medicine, Houston, Texas
Introduction
Heart failure (HF) is a prevalent and serious condition associated with substantial morbidity and mortality. Management strategies have evolved, with recent focus on sodium-glucose cotransporter 2 (SGLT2) inhibitors potentially offering novel therapeutic benefits.
SGLT2 Inhibitors in HF with Reduced Ejection Fraction (HFrEF)
Studies have demonstrated that dapagliflozin, an SGLT2 inhibitor, significantly reduces the risk of worsening HF or cardiovascular death in patients with HFrEF. (1,3) The DAPA-HF trial, a pivotal phase 3 study, enrolled patients with New York Heart Association (NYHA) class II-IV symptoms and ejection fractions ≤ 40%. Results showed a reduction in the primary outcome (composite of worsening HF or cardiovascular death) among those treated with dapagliflozin compared to placebo (HR 0.74, 95% CI 0.65-0.85). This benefit was observed regardless of diabetes status, highlighting potential glucose-independent mechanisms of action.
SGLT2 Inhibitors in HF with Mildly Reduced or Preserved Ejection Fraction
Emerging evidence suggests that dapagliflozin may also benefit patients with HF and preserved or mildly reduced ejection fraction (HFpEF and HFmrEF). (2) The DELIVER trial randomized patients with HF and ejection fractions > 40% to dapagliflozin or placebo. Results indicated a reduction in the composite of worsening HF or cardiovascular death with dapagliflozin (HR 0.82, 95% CI 0.73-0.92). Moreover, improvements in symptom burden were noted, supporting potential broad utility across HF phenotypes.
Mechanisms of Action
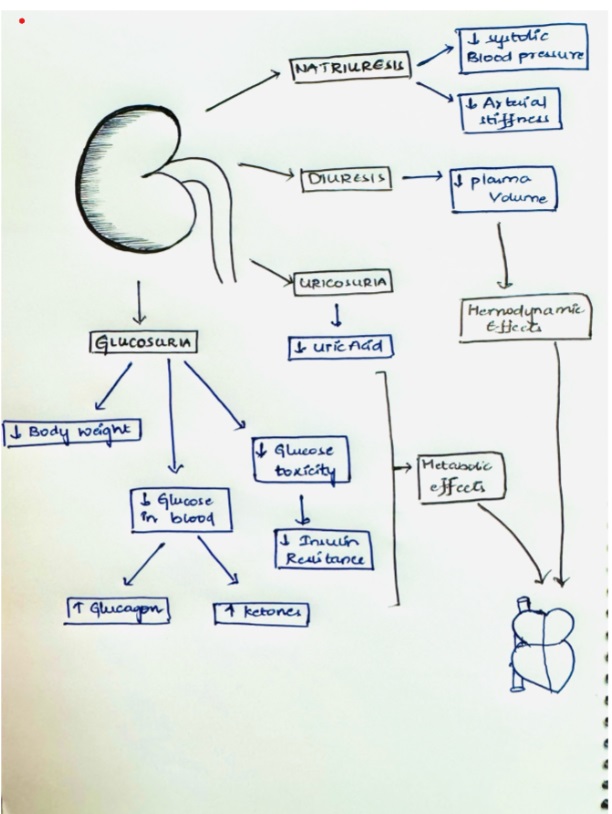
Beyond glycemic control, SGLT2 inhibitors like dapagliflozin may exert beneficial effects in HF through mechanisms including improved cardiac remodeling, reduced sodium retention, decrease plasma volume, and potential metabolic modulation. These mechanisms contribute to improved hemodynamics and reduced cardiovascular morbidity and mortality, as observed in clinical trials.
Contraindication
eGFR < 30mL/minute/1.73 m2
Safety and Tolerability
Safety profiles across trials indicate that dapagliflozin is generally well tolerated in HF patients, with adverse event rates comparable to placebo. Common side effects include volume depletion and renal dysfunction (Acute kidney injury due to dehydration), which are manageable with appropriate monitoring. Other severe side effects are euglycemic diabetic ketoacidosis, allergic reaction and genital infections (mostly fungal).
Canagliflozin increase the risk of lower limb amputation and bone fractures.
Clinical Implications and Future Directions
The efficacy of SGLT2 inhibitors in reducing HF-related events underscores their potential as adjunctive therapy in standard HF management. Future research should focus on optimizing patient selection, exploring combination therapies, and elucidating long-term benefits and risks in diverse patient populations.
Conclusion
SGLT2 inhibitors like dapagliflozin show great promise in improving outcomes for patients with heart failure. These medications not only reduce cardiovascular risks but also improve quality of life, highlighting their potential as essential elements of comprehensive heart failure treatment strategies.
References
McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukát A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjöstrand M, Langkilde AM; DAPA-HF Trial Committees and Investigators. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2019 Nov 21;381(21):1995-2008. doi: 10.1056/NEJMoa1911303. Epub 2019 Sep 19. PMID: 31535829.
Solomon SD, McMurray JJV, Claggett B, de Boer RA, DeMets D, Hernandez AF, Inzucchi SE, Kosiborod MN, Lam CSP, Martinez F, Shah SJ, Desai AS, Jhund PS, Belohlavek J, Chiang CE, Borleffs CJW, Comin-Colet J, Dobreanu D, Drozdz J, Fang JC, Alcocer-Gamba MA, Al Habeeb W, Han Y, Cabrera Honorio JW, Janssens SP, Katova T, Kitakaze M, Merkely B, O’Meara E, Saraiva JFK, Tereshchenko SN, Thierer J, Vaduganathan M, Vardeny O, Verma S, Pham VN, Wilderäng U, Zaozerska N, Bachus E, Lindholm D, Petersson M, Langkilde AM; DELIVER Trial Committees and Investigators. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med. 2022 Sep 22;387(12):1089-1098. doi: 10.1056/NEJMoa2206286. Epub 2022 Aug 27. PMID: 36027570.
Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, Khariton Y, Malik AO, Khumri T, Umpierrez G, Lamba S, Sharma K, Khan SS, Chandra L, Gordon RA, Ryan JJ, Chaudhry SP, Joseph SM, Chow CH, Kanwar MK, Pursley M, Siraj ES, Lewis GD, Clemson BS, Fong M, Kosiborod MN. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med. 2021 Nov;27(11):1954-1960. doi: 10.1038/s41591-021-01536-x. Epub 2021 Oct 28. PMID: 34711976; PMCID: PMC8604725.
Petrie MC, Verma S, Docherty KF, Inzucchi SE, Anand I, Belohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukát A, Ge J, Howlett J, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Vinh PN, Schou M, Tereshchenko S, Køber L, Kosiborod MN, Langkilde AM, Martinez FA, Ponikowski P, Sabatine MS, Sjöstrand M, Solomon SD, Johanson P, Greasley PJ, Boulton D, Bengtsson O, Jhund PS, McMurray JJV. Effect of Dapagliflozin on Worsening Heart Failure and Cardiovascular Death in Patients With Heart Failure With and Without Diabetes. JAMA. 2020 Apr 14;323(14):1353-1368. doi: 10.1001/jama.2020.1906. Erratum in: JAMA. 2021 Apr 6;325(13):1335. doi: 10.1001/jama.2021.2802. PMID: 32219386; PMCID: PMC7157181.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS; DECLARE–TIMI 58 Investigators. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2019 Jan 24;380(4):347-357. doi: 10.1056/NEJMoa1812389. Epub 2018 Nov 10. PMID: 30415602.
Lipoprotein(a) is a variation of low-density lipoprotein that includes a protein known as apolipoprotein(a). Research involving genetics and population studies has found that elevated levels of lipoprotein(a) is associated with an increased risk of developing atherosclerosis and related conditions like coronary heart disease and stroke.(1)
1.2. Structure of the Lipoprotein (a) particle
Lp(a) shares similarities with low-density lipoprotein (LDL), as it consists of a lipid core containing cholesteryl esters and triacylglycerols, surrounded by a phospholipid and apolipoprotein B-100 (apoB-100) particle layer.(3) In contrast to LDL cholesterol, Lp(a) contains an additional unique glycoprotein known as apolipoprotein(a) (apo(a)), which is connected to apoB-100 through a single disulfide bond. The biosynthesis of Lipoprotein(a) takes place almost exclusively in the liver.
2.1. Adverse effects of elevated lipoprotein (a) levels
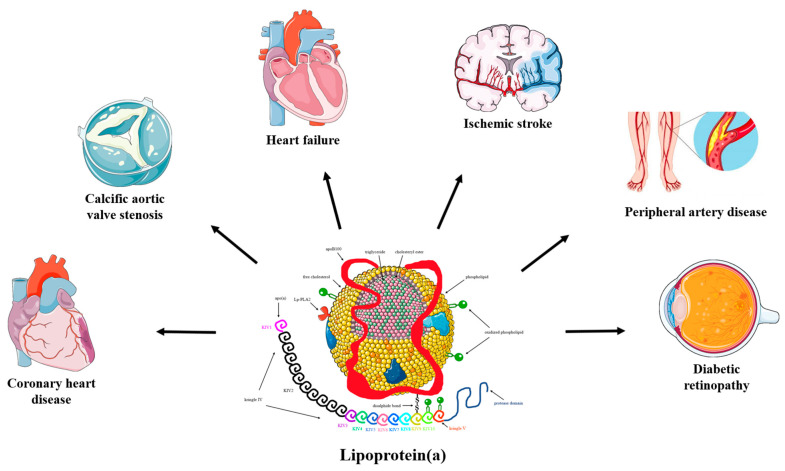
Similar to LDL-cholesterol, the cholesterol found in Lp(a) can accumulate in the walls of your blood vessels. The higher your Lp(a) level, the greater the likelihood of this occurring. These deposits of cholesterol, known as plaques, have the potential to reduce blood flow to various organs such as the heart, brain, kidneys, lungs, legs, and other parts of the body. Over time, plaques can gradually grow or suddenly rupture, obstructing blood vessels and resulting in heart attacks or strokes.Lp(a) has the potential to induce heightened clotting, which in turn can result in the formation of blockages in blood vessels at a rapid pace. Lp(a) promotes inflammation which increases the likelihood that plaques will rupture.(2)Additionally, heightened levels of Lp(a) can contribute to the development of aortic stenosis, a condition characterized by the narrowing of the aortic valve due to inflammation. This chronic inflammation can lead to the accumulation of calcium on the valve, resulting in stiffness. As a consequence, blood flow may be compromised if the valve fails to open fully. In certain instances, individuals with aortic stenosis may require surgical intervention or a procedure to replace the affected aortic valve.(2) Convincing evidence has emerged from pathophysiological, epidemiological, and genetic studies on the causality of high serum lipoprotein(a) (Lp(a)) levels as a potent risk factor for coronary heart disease (CHD), ischemic stroke, peripheral artery disease, heart failure, calcific aortic valve stenosis (CAVS), mitral valve stenosis, and retinopathy in patients with diabetes.(14) (Fig. 1)
Fig. 1: The pathogenicity of lipoprotein(a). (Source: Koutsogianni et al, 2023)
Elevated levels of Lp(a), specifically exceeding 50 mg/dL (125 nmol/L), are prevalent. The median levels of Lp(a) differ according to race and gender. This condition is observed in individuals of all races and ethnicities, but it appears to be more prevalent among black individuals. Many individuals with high Lp(a) do not experience any symptoms. However, if you possess the following risk factors, your doctor may suspect that you have high Lp(a).
Poor circulation in your legs (called peripheral arterial disease)
Heart attack, stroke, or coronary artery disease before age 55 (in men) or age 65 (in women) without known risk factors, such as high LDL, smoking, diabetes, or obesity
Female family members who had a heart attack or stroke before age 65
Male family members who had a heart attack or stroke before age 55
Familial hypercholesterolemia
Aortic valve stenosis
2.2. Mechanism of action
Lp(a) favors initiation of atherogenesis by modulating recruitment of inflammatory cells in the vessel wall. In vitro and animal studies have implicated lipoprotein(a) in key processes in atherosclerosis, including foam cell formation, smooth muscle cell proliferation, and plaque inflammation and instability.(4) Lp(a) blocks plasminogen conversion to plasmin, therefore plasmin mediated TGF-β activation is inhibited. TGF-β is an autocrine inhibitor of Smooth muscle cell growth.(12) Lp(a) increases atherosclerotic plaque vulnerability, vascular smooth muscle cell proliferation and adhesion of molecules, chemotactic factors and plasma cytokines. (Fig. 2) Moreover, Lp(a) enhances platelet activation and aggregation and inhibits fibrinolysis by inhibiting plasminogen activation.(13)
Fig. 2: The impact of lipoprotein(a) on atherosclerotic process and atherothrombosis. (source:Lampsas et al, 2023)
Recently, lipoprotein(a) has also been identified as the main carrier of oxidized phospholipids considered proinflammatory and proatherogenic.(5) Lipoprotein(a) has been hypothesized to contribute to wound healing (6), transporting cholesterol to sites of injury for cell replenishment, and limiting bleeding via attenuated fibrinolysis. A nonspecific wound healing effect of lipoprotein(a) may, however, explain an association with aortic valve disease considered the result of repeated valve injury and repair mechanisms (7). Recent in vitro studies, demonstrating osteogenic differentiation of valvular interstitial cells exposed to lipoprotein(a) and associated oxidized phospholipids, point to yet another possible mechanism relevant for aortic valve disease, often characterized by pronounced valve calcification (8), and perhaps also for development of advanced atherosclerotic lesions. Large genetic epidemiologic studies have generated renewed interest in lipoprotein(a) by providing strong genetic evidence of causal associations of high lipoprotein(a) concentrations with increased risk of CHD, AVS, heart failure, and mortality.(9,10,11)
3. Treatment options in high lipoprotein(a)
Lipoprotein apheresis
The most effective clinical intervention available for lowering Lp(a) is lipoprotein apheresis (LA). Following treatment, Lp(a) levels can be acutely reduced by 70–80%. However, rebound elevations between apheresis sessions, carried out weekly, biweekly, or less frequently, typically yield a mean Lp(a) reduction of 25–40%, depending on the treatment duration and initial Lp(a) levels. Although LA is not commonly utilized worldwide, except in Germany, long-term studies on patients with elevated Lp(a) undergoing LA indicate that this therapy may potentially reduce 5-year cardiovascular risk by up to 86%.(15)
Niacin
Niacin decreases Lp(a) in a dose-dependent manner by approximately 30–40% on average, but only by 18% in those with the highest Lp(a) levels. The effect of niacin is likely due to a decreased apo(a) production rate. Importantly, studies with cardiovascular outcomes showed no benefit of adding niacin to statins. Moreover, niacin use is limited by side effects, such as flushing, gastrointestinal discomfort, and new-onset diabetes. (14)
PCSK9 inhibitors
PCSK9 monoclonal antibodies, i.e., alirocumab and evolocumab, reduce Lp(a) levels by approximately 20–30%. The exact mechanism by which PCSK9 inhibitors reduce Lp(a) levels remains unclear, Current hypotheses include increased clearance of Lp(a) particles via the LDLR, increased clearance of Lp(a) via other receptors (the LDL receptor-related protein 1, the cluster of differentiation 36 receptor, toll-like receptor 2, scavenger receptor-B1, and plasminogen receptors), as well as a reduction in apo(a) production, secretion, and/or assembly.(14)
Fibrates
Bezafibrate has been shown to reduce Lp(a) levels by approximately 13–39%. (16)
Lomitapide
Lomitapide, a microsomal triglyceride transfer protein inhibitor, reduces Lp(a) levels by 15–19%(17) The possible mechanism for Lp(a)-lowering is the decrease in very-low-density lipoprotein (VLDL) and chylomicron synthesis via inhibition of MTP. MTP is located in the endoplasmic reticulum of hepatocytes and enterocytes and is most likely responsible for transferring triglycerides to nascent apoB as it enters the lumen of the endoplasmic lumen. Consequently, MTP inhibition seems to control the number of apoB-containing lipoprotein particles secreted into the bloodstream, including Lp(a) particles.(17)
Cholesteryl Transfer Protein (CETP) Inhibitors
CETP mediates the transfer of cholesteryl esters from high-density lipoprotein (HDL) to apoB100-containing particles, including VLDL and LDL, in exchange for triglycerides. Apart from raising HDL cholesterol, CETP inhibitors also decrease apoB100, LDL cholesterol, and Lp(a) levels (by approximately 24–36%). (14) Clinical outcome trials of four CETP inhibitors have been completed, including torcetrapib, dalcetrapib, evacetrapib, and anacetrapib. CETP inhibitors are not currently approved for clinical use.
Conclusions:
Lp(a) has been identified as a significant risk factor for atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve stenosis (CAVS), with the risk increasing in proportion to Lp(a) concentrations. It is believed that meaningful reductions in cardiovascular outcomes require substantial reductions in Lp(a) concentrations. In patients with elevated Lp(a) levels, more aggressive targets for LDL cholesterol (LDL-C) reduction are recommended compared to individuals with similar ASCVD risk but normal Lp(a) levels. Initially, the approach to reducing ASCVD risk in patients with high Lp(a) focuses on further LDL-C reduction. However, there are several promising agents in late-stage development, and the results of phase 3 outcome studies are highly anticipated. Additionally, there are other agents in various drug classes, including lipid-modifying medications, that can also impact Lp(a) levels.
References:
1: Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010
3. Ruscica M., Sirtori C.R., Corsini A., Watts G.F., Sahebkar A. Lipoprotein(a): Knowns, unknowns and uncertainties. Pharmacol. Res. 2021;173:105812. doi: 10.1016/j.phrs.2021.105812.
4. Boffa MB , Marcovina SM , Koschinsky ML. Lipoprotein(a) as a risk factor for atherosclerosis and thrombosis: mechanistic insights from animal models. Clin Biochem 2004;37:333–43.
5. Tsimikas S , Brilakis ES , Miller ER , McConnell JP , Lennon RJ , Kornman KS , et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med 2005;353:46–57.
6. Brown MS , Goldstein JL. Plasma lipoproteins: teaching old dogmas new tricks. Nature 1987;330:113–4.
7. Kamstrup PR , Tybjærg-Hansen A , Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol 2014;63:470–7.
8. Zheng KH , Tsimikas S , Pawade T , Kroon J , Jenkins WSA , Doris MK , et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol 2019;73:2150–62.
9. Thanassoulis G , Campbell CY , Owens DS , Smith JG , Smith AV , Peloso GM , et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med 2013;368:503–12.
10. Kamstrup PR , Tybjærg-Hansen A , Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol 2014;63:470–7.
11. Arsenault BJ , Boekholdt SM , Dubé M-P , Rhéaume É , Wareham NJ , Khaw K-T , et al. Lipoprotein(a) levels, genotype and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet 2014;7:304–10.
12 Grainger D.J., Kirschenlohr H.L., Metcalfe J.C., Weissberg P.L., Wade D.P., Lawn R.M. Proliferation of human smooth muscle cells promoted by lipoprotein(a) Science. 1993;260:1655–1658.
13. Lampsas S, Xenou M, Oikonomou E, Pantelidis P, Lysandrou A, Sarantos S, Goliopoulou A, Kalogeras K, Tsigkou V, Kalpis A, Paschou SA, Theofilis P, Vavuranakis M, Tousoulis D, Siasos G. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules. 2023 Jan 18;28(3):969.
14. Koutsogianni AD, Liamis G, Liberopoulos E, Adamidis PS, Florentin M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels-Potent Clinical Implications. Pharmaceuticals (Basel). 2023 May 16;16(5):750.
15. Roeseler E., Julius U., Heigl F., Spitthoever R., Heutling D., Breitenberger P., Leebmann J., Lehmacher W., Kamstrup P.R., Nordestgaard B.G., et al. Lipoprotein apheresis for lipoprotein(a)-Associated cardiovascular disease: Prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler. Thromb. Vasc. Biol. 2016;36:2019–2027.
16. Tenenbaum A., Fisman E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012;11:140. doi: 10.1186/1475-2840-11-140.
17. Cuchel M., Meagher E.A., Theron H.D.T., Blom D.J., Marais A.D., Hegele R.A., Averna M.R., Sirtori C.R., Shah P.K., Gaudet D., et al. Efficacy and Safety of a Microsomal Triglyceride Transfer Protein Inhibitor in Homozygous Familial Hypercholesterolemia. Lancet. 2013;381:40.