Authors: Tejaswini Potlabathini, Elaine Tran, PA Student, University of Texas Medical Branch, Galveston, Tx. Amin H. Karim MD FRCP FACC
Patient Overview
A 72-year-old female with a past medical history of hypertension, hyperlipidemia, hypothyroidism, and prediabetes presented to the cardiology clinic for a routine follow-up. On examination, she was found to have a firm, nodular thickening of tissue on one palm over the third metacarpal area, associated with nearby skin puckering, prompting a presumptive diagnosis of Dupuytren’s contracture.
Description
Dupuytren’s contracture is a progressive fibroproliferative disorder of the palmar fascia that leads to flexion contractures of the digits. It affects approximately 0.6% to 31.6% of the general population, with prevalence increasing with age. While its exact cause remains unclear, it is believed to have a multifactorial etiology involving both genetic and environmental factors. Genetically, it follows an autosomal dominant inheritance pattern more commonly in individuals of Northern European descent. The condition predominantly affects men but can also occur in women, often with a later onset and milder progression. Dupuytren’s contracture is associated with other fibrotic disorders, including plantar fibromatosis (Ledderhose disease) and dorsal hand nodules (Garrod pads). Several risk factors have been identified, including diabetes mellitus, smoking, and chronic alcohol consumption, all of which may contribute to microvascular ischemia and tissue damage. Additionally, the condition has been linked to seizure disorders and chronic liver disease, particularly in patients with cirrhosis. Occupational risk factors, such as repetitive manual labor and prolonged exposure to hand-transmitted vibration, may also accelerate disease progression.
Etiology
Fibroblasts, which are mesenchymal cells responsible for tissue maintenance, play a crucial role in the pathology of Dupuytren’s contracture. The disease progresses through three stages: proliferative, involutional, and residual. In the proliferative stage, fibroblasts are stimulated and differentiate into mature myofibroblasts under the influence of transforming growth factor-beta (TGF-β) and mechanical stress from associated risk factors. As the disease advances to the involutional stage, nodules begin to form, producing an extracellular matrix (ECM) rich in type III collagen. In the residual stage, fibrotic tissue stabilizes as the ratio of type I to type III collagen increases, leading to collagen cross-linking. This results in the formation of fibrous cords, which cause progressive digital flexion contractures. Additionally, the presence of CD3-positive lymphocytes and the expression of major histocompatibility complex (MHC) class II proteins suggest a possible role for a T-cell-mediated autoimmune response in the disorder. In summary, Dupuytren’s contracture arises from fibroblastic proliferation and disorganized collagen deposition, ultimately leading to palmar fascial thickening and contracture formation.
Patients with Dupuytren’s contracture may present with painful or painless lumps in the palm, along with restricted finger mobility and decreased grip strength. As the nodules thicken and fibrous cords form, patients may experience difficulty straightening or spreading their fingers due to flexion contractures. The fourth and fifth digits are most commonly affected, with contractures typically involving the metacarpophalangeal (MCP) joint first, followed by the proximal interphalangeal (PIP) joint, and less frequently, the distal interphalangeal (DIP) joint.
Physical Examination
On physical examination, the Hueston Tabletop Test can be performed. Patients are asked to place their palm flat on a table, and failure to do so indicates a positive test. If contractures are present, the angles at the MCP and PIP joints should be measured to assess disease severity and progression. Grading of Dupuytren’s contracture is as follows: Grade 1 presents as a thickened nodule and band in the palmar aponeurosis, which may progress to skin tethering, puckering, or pitting. Grade 2 presents as a peritendinous band, leading to limited extension of the affected finger. Grade 3 presents as a significant flexion contracture.
Treatment
Although there is no definitive cure for Dupuytren’s contracture, symptoms can be managed through nonsurgical and surgical options. Nonsurgical options include corticosteroid injections, collagenase clostridium histolyticum injections, and needle aponeurotomy. Corticosteroids alleviates symptoms by reducing inflammation while targeted collagenase injections can target and enzymatically degrade the collagen. In needle aponeurotomy, a fine needle is used to precisely cut through the fibrous cord. While this minimally invasive procedure does not remove the cord, a break in the cord allows for improved finger motion. Although more invasive and extensive, a partial palmar fasciectomy removes the abnormal tissue fibrous tissue and cords. Postoperative care includes splinting, wound care, and physical therapy. Emerging therapies include use of anti-tumor necrosis factors such as adalimumab injections to slow disease progression by targeting inflammatory pathways.
Hindocha S, Stanley JK, Watson S, Bayat A. Revised Tubiana’s staging system for assessment of disease severity in Dupuytren’s disease—preliminary clinical findings. EBioMedicine. 2018;36:86-90. doi:10.1016/j.ebiom.2018.06.022.
Zarb RM, Graf AR, Talhelm JE, et al. Dupuytren’s contracture recurrence and treatment following collagenase Clostridium histolyticum injection: a longitudinal assessment in a veteran population. Mil Med. 2023;188(9-10):e2975-e2981. doi:10.1093/milmed/usad075.
Hema Manvi Koneru, MBBS Rajiv Gandhi Institute of Medical Sciences, Telangana, India.
Amanpreet Kaur, MBBS Government Medical College Patiala, India.
Divyasri Koneru, MBBS Dr.Pinnamaneni Siddhartha Institute of Medical Sciences and Research Foundation, India
Amin H. Karim MD FACC, Clinical assistant professor, Baylor College of Medicine, Houston, Texas.
A 74-year-old male presented with complaints of dizziness. His medical history includes hypertension, diabetes mellitus, mitral regurgitation, tricuspid regurgitation, and a transient ischemic attack five months prior. Additionally, he reported two episodes of memory lapses within the past year.
The patient denied experiencing orthopnea, paroxysmal nocturnal dyspnea, chest pain, smoking, shortness of breath, leg swelling, speech disturbances, disequilibrium, blurry vision, syncope, tinnitus, hearing loss, ataxia, numbness, tingling, pins, and needles in the arms or legs.
MEDICATIONS
Aspirin 81mg sustained-release oral tablet, once daily; atorvastatin calcium 80mg tablet, taken orally once daily; clopidogrel 75mg tablet, taken orally daily; dulaglutide 0.75mg/0.5ml subcutaneous pen injector, administered subcutaneously once weekly; gabapentin 300mg oral capsule, taken once daily; losartan 25mg tablet, taken orally once daily; metformin hydrochloride 750mg extended-release tablet, taken twice daily with meals; nitroglycerin 0.4 mg sublingual tablet, to be used when chest pain persists; and thiamine 100 mg oral tablet, taken once daily. These medications have been prescribed to effectively manage the patient’s medical conditions and symptoms.
The patient had a history of allergy to the contrast medium used for the radiological examination.
LAB WORKUP
HBA1C-6.6gm/dl
The rest of the blood reports- are within in normal range.
ELECTROCARDIOGRAM
Sinus tachycardia with nonspecific ST-abnormality
CTA CORONARY ARTERIES
There is severe coronary calcification. The observed calcium score is 780, which is at the 75th percentile for subjects of the same age, gender, race/ethnicity who are free of clinical cardiovascular disease and treated diabetes.
Moderate to severe, predominantly calcified. Coronary artery disease involving proximal and LAD with moderate luminal stenosis. FFR-CT was performed on the LAD which showed significant flow limitation of the mid-LAD (left anterior descending artery).
CTA OF BRAIN AND NECK
Brain–moderate chronic microvascular ischemia, suprasellar 8 x 7 mm partially calcified lipoma or dermoid cyst.
Neck-Complete occlusion of the left vertebral artery V1 and V2 segments. Atherosclerosis of bilateral extracranial carotid arteries without significant stenosis.
The head shows no significant stenosis or occlusion.
MRI BRAIN
No acute ischemia.
Hypothalamus showing non-enhancing mass suggestive of Lipoma/Dermoid cyst.
Moderate chronic microvascular ischemia.
ECHOCARDIOGRAM
Mild left ventricular hypertrophy.
Trace tricuspid regurgitation.
Mitral regurgitation.
Left ventricular dysfunction.
NUCLEAR STRESS TEST
A small area of perfusion defect in the inferior wall which fills with reperfusion
CAROTID ULTRASOUND
No occlusive disease.
Following a thorough workup and assessment of the patient’s clinical presentation, imaging studies, and diagnostic tests, a final diagnosis of subclavian steal syndrome was made. This condition is attributed to the underlying pathology of atherosclerosis which mainly affects the subclavian artery.
DEFINITION
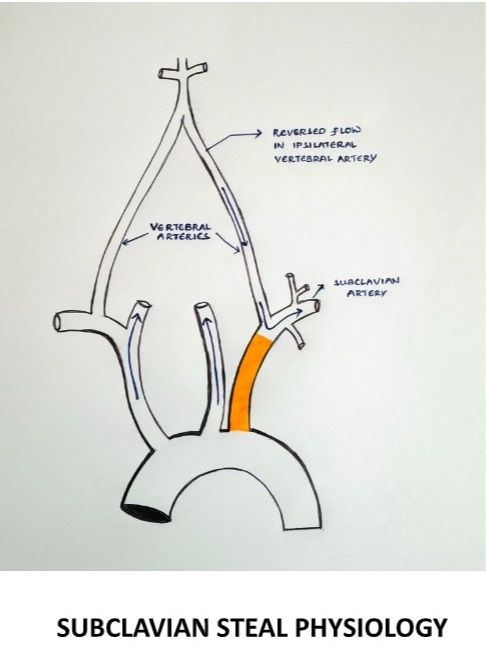
It is characterized by the retrograde flow of blood in the vertebral artery, primarily resulting from significant stenosis or occlusion in the pre-vertebral subclavian artery resulting in cerebrovascular symptoms on the ipsilateral side of the occlusion.
§ During exertion, the subclavian artery steals blood from the vertebrobasilar artery circulation to supply the arm, which leads to vertebrobasilar insufficiency.
SUBCLAVIAN STEAL PHYSIOLOGY
EPIDEMIOLOGY
The prevalence of subclavian steal syndrome has not been well defined, although subclavian steal physiology has been reported in 1.3 to 2.6% of the general population of patients with extracranial atherosclerosis (1,2) as well as within subsets of those presenting with acute ischemic stroke (3)
ETIOLOGY
The most common cause is atherosclerosis.
Other vascular causes- Takayasu arteritis, giant cell arteritis
Arterial thoracic outlet syndrome
Congenital anomalies- anomalies of the aortic arch, anomalies of the brachiocephalic trunk
A consequence of corrective surgery- surgical repair of the Tetralogy of Fallot with a Blalock-Taussig anastomosis, surgical management of the coarctation of the aorta.
Subclavian stenosis more commonly occurs on the left side (>75 percent), possibly due to more acute origin on the left subclavian artery resulting in accelerated atherosclerosis from increased turbulence. (4,5,6)
CLINICAL FEATURES
Neurovascular symptoms can be caused by vertebrobasilar ischemia of the brainstem or cerebellum
Symptoms include dizziness, vertigo, binocular double vision, dysarthria, syncope, and drop attacks (sudden falls without loss of consciousness) (7)
Episodes can be precipitated by exercise of the ischemic arm and precipitated by certain head movements.
Atypical neurological presentations- there is also increasing recognition that patients with vertebrobasilar insufficiency can present with other types of symptoms not directly attributable to brainstem or cerebellar ischemia.
Subarachnoid hemorrhage (8)
Cognitive and mood abnormalities
EVALUATION
Although neurovascular symptoms associated with subclavian steal physiology occur in only a minority of patients, symptoms of vertebrobasilar ischemia demand critical evaluation (10,11)
When to suspect subclavian steal syndrome
Diagnosis should be considered in patients with a measurable upper extremity blood pressure differential who develop episodic neurological symptoms attributable to the brainstem or cerebellar ischemia.
Suspicion is increased if the episodes are provoked by arm exercise, especially if accompanied by ischemic arm symptoms (7)
PHYSICAL EXAMINATION
All major pulses should be palpated, and blood pressure should be checked in both arms.
It is important to examine the subclavian arteries in the supraclavicular fossa using palpation and auscultation for paraventricular bruits, also vertebral and carotid arteries for the evidence of any occlusive arterial disease.
On examination, there may be a significant decrease in the blood pressure on the affected side with a pulse delay appreciated when palpating the radial arteries simultaneously known as RADIO-RADIAL DELAY.
Check for any evidence of thromboembolism in the skin of the hands and nailbeds of the affected extremities.
Ischemia affecting the temporo-occipital areas of the cerebral hemispheres or segments of the brainstem and cerebellum characteristically produces bilateral symptoms.
RADIOLOGICAL EXAMINATION
Initial vascular imaging
1. Duplex ultrasound
A subclavian artery peak systolic velocity >240cm/sec is predictive of a significant (>70 percent) subclavian artery stenosis (9)
When severe stenosis (>80 percent narrowing) of the proximal subclavian artery is present, 65 percent of pa>ents have permanent flow reversal in the ipsilateral vertebral artery, and 30 percent have intermittent flow reversal (10,11)
2. Transcranial doppler ultrasound
Confirmatory vascular imaging
Computed tomographic angiography (CTA)-provides precise measurement of the severity of the stenosis. main drawback is the absence of dynamic flow information.
Magnetic resonance angiography (MRA)- sensitive than CTA. Does not depict images of arterial anatomy but rather the behavior and speed of the flowing protons in the vessel. Also provides information on the intracranial cerebrovascular circulation.
Catheter-based digital subtraction angiography (DSA)
DIAGNOSIS
DIAGNOSTIC CRITERIA– the presence of subclavian steal physiology with demonstration of a subclavian artery stenosis/ occlusion proximal to the origin of the vertebral artery causing
Marked reduction in the ipsilateral brachial artery blood pressure.
Reversal of the direction of blood flow in the ipsilateral vertebral artery
Neurological symptoms referrable to the vertebrobasilar circulation (cerebellum, brainstem, thalamus, occipital regions)
The likelihood that the presence of subclavian steal physiology will lead to the subclavian steal syndrome increases as the brachial pressure differential between the two limbs becomes more pronounced, particularly >40mmhg (5)
DIFFERENTIAL DIAGNOSIS
These include all the potential causes of vertebrobasilar embolism due to atherosclerosis, hypertension, hypercoagulable states, tumors, tobacco smoking, trauma, and others
MANAGEMENT
Management of patients with subclavian steal syndrome is individualized depending on the etiology, type, and severity of symptoms and their impact on quality of life
ATHEROSCLEROTIC SUBCLAVIAN STEAL SYNDROMEAPPROACH
For most patients with atherosclerosis as the etiology for subclavian steal syndrome, conservative management is the preferred initial therapy.
Initial medical management
Start with the low-dose aspirin.
However, the addition of other anti-platelet drugs does not seem justified since this condition is hemodynamic derangement. once a decision is made for surgical management, additional anti-thrombotic agents are required.
Optimal blood pressure management is required within the ipsilateral side brachial artery.
Surgical management
Proximal subclavian endarterectomy, which is a trans-thoracic approach used for revascularizing the subclavian artery.
Re-vascularization surgery via endovascular techniques (carotid-subclavian bypass or subclavian-carotid transposition).
Carotid intervention in patients with severe concomitant carotid artery disease helps to improve cerebral perfusion.
NON-ATHEROSCLEROTIC SUBCLAVIAN STEAL SYNDROME
For non-atherosclerotic subclavian syndrome etiologies, the primary cause needs to be addressed.
Example- Takayasu arteritis is by systemic glucocorticoids or steroid-sparing agents like biological DMARD (Disease Modifying Anti-Rheumatoid Drugs) or non-biological DMARD
CONCLUSION
This case highlights the importance of a detailed evaluation of subclavian steal syndrome can be due to atherosclerosis or non-atherosclerotic causes like Takayasu arteritis, giant cell arteritis, arterial thoracic outlet syndrome, and other causes. In our case, it might be a subclavian steal syndrome secondary to atherosclerosis.
REFERENCES
Fields WS, Lemak NA. Joint Study of extracranial arterial occlusion. VII. Subclavian steal–a review of 168 cases. JAMA. 1972 Nov 27;222(9):1139-43. PMID: 4678043.
Hennerici M, Klemm C, Rautenberg W. The subclavian steal phenomenon: a common vascular disorder with rare neurologic deficits. Neurology. 1988 May;38(5):669-73. doi: 10.1212/wnl.38.5.669. PMID: 3362359.
Bajko Z, Motataianu A, Stoian A, Barcutean L, Andone S, Maier S, Drăghici IA, Cioban A, Balasa R. Prevalence and Clinical Characteristics of Subclavian Steal Phenomenon/Syndrome in Patients with Acute Ischemic Stroke. J Clin Med. 2021 Nov 10;10(22):5237. doi: 10.3390/jcm10225237. PMID: 34830519; PMCID: PMC8621575.
Ochoa VM, Yeghiazarians Y. Subclavian artery stenosis: a review for the vascular medicine practitioner. Vasc Med. 2011 Feb;16(1):29-34. doi: 10.1177/1358863X10384174. Epub 2010 Nov 15. PMID: 21078767.
Labropoulos N, Nandivada P, Bekelis K. Prevalence and impact of the subclavian steal syndrome. Ann Surg. 2010 Jul;252(1):166-70. doi: 10.1097/SLA.0b013e3181e3375a. PMID: 20531004.
Shadman R, Criqui MH, Bundens WP, Fronek A, Denenberg JO, Gamst AC, McDermott MM. Subclavian artery stenosis: prevalence, risk factors, and association with cardiovascular diseases. J Am Coll Cardiol. 2004 Aug 4;44(3):618-23. doi: 10.1016/j.jacc.2004.04.044. PMID: 15358030.
KESTELOOT H, VANHOUTE O. REVERSED CIRCULATION THROUGH THE VERTEBRAL ARTERY. Acta Cardiol. 1963;18:285-99. PMID: 14045892.
Rodriguez-Lopez JA, Werner A, Martinez R, Torruella LJ, Ray LI, Diethrich EB. Stenting for atherosclerotic occlusive disease of the subclavian artery. Ann Vasc Surg. 1999 May;13(3):254-60. doi: 10.1007/s100169900254. PMID: 10347257.
Gutierrez GR, Mahrer P, Aharonian V, Mansukhani P, Bruss J. Prevalence of subclavian artery stenosis in patients with peripheral vascular disease. Angiology. 2001 Mar;52(3):189-94. doi: 10.1177/000331970105200305. PMID: 11269782.
Osiro S, Zurada A, Gielecki J, Shoja MM, Tubbs RS, Loukas M. A review of subclavian steal syndrome with clinical correlation. Med Sci Monit. 2012 May;18(5):RA57-63. doi: 10.12659/msm.882721. PMID: 22534720; PMCID: PMC3560638.
Cornelissen SA, Heye S, Maleux G, Daenens K, van Loon J, De Vleeschouwer S. Treatment of ruptured subclavian steal flow-related vertebrobasilar junction aneurysms: Case report on surgical and endovascular considerations from two cases. Int J Surg Case Rep. 2022 Jan;90:106744. doi: 10.1016/j.ijscr.2021.106744. Epub 2021 Dec 30. PMID: 34991048; PMCID: PMC8741505.
Ahuja CK, Joshi M, Mohindra S, Khandelwal N. Vertebrobasilar Junction Aneurysm Associated with Subclavian Steal: Yet another Hemodynamic Cause for Aneurysm Development and Associated Challenges. Neurol India. 2020 May-Jun;68(3):708-709. doi: 10.4103/0028-3886.288981. PMID: 32643699.
Tonetti DA, Jankowitz BT. Subclavian Steal Flow-Related Aneurysm Formation. World Neurosurg. 2019 May;125:101-103. doi: 10.1016/j.wneu.2019.01.186. Epub 2019 Feb 8. PMID: 30743034.
Mousa AY, Morkous R, Broce M, Yacoub M, Sticco A, Viradia R, Bates MC, AbuRahma AF. Validation of subclavian duplex velocity criteria to grade severity of subclavian artery stenosis. J Vasc Surg. 2017 Jun;65(6):1779-1785. doi: 10.1016/j.jvs.2016.12.098. Epub 2017 Feb 17. PMID: 28222983.
Nicholls SC, Koutlas TC, Strandness DE. Clinical significance of retrograde flow in the vertebral artery. Ann Vasc Surg. 1991 Jul;5(4):331-6. doi: 10.1007/BF02015293. PMID: 1878290.
Harper C, Cardullo PA, Weyman AK, Patterson RB. Transcranial Doppler ultrasonography of the basilar artery in patients with retrograde vertebral artery flow. J Vasc Surg. 2008 Oct;48(4):859-64. doi: 10.1016/j.jvs.2008.05.057. Epub 2008 Aug 9. PMID: 18692344.
By Edward Aldrich Medical Officer, MBCHB Stellenbosch University, South Africa
Amin H. Karim MD Baylor College Of Medicine, Methodist Academic Institute. Houston, Texas
Patient is a 70-year-old African American male known with atrial fibrillation, hypertension, GERD, venous insufficiency, sleep apnea and benign prostatic hyperplasia, who presented with heart failure due to hypertrophic cardiomyopathy. The diagnosis of transthyretin cardiac amyloidosis was confirmed on biopsy, after suggestive features were seen on echocardiogram and cardiac magnetic resonance.
Epidemiology – Cardiac amyloidosis is a rare form of cardiomyopathy and approximately 95 percent of cases are caused by the deposition of transthyretin (ATTR amyloidosis) or immunoglobulin light chains (AL amyloidosis).
Classification of amyloidosis is based upon the type of precursor protein:
Transthyretin amyloidosis (ATTR amyloidosis) – Transthyretin amyloidosis results from the misfolding and deposition of transthyretin (TTR, formerly known as prealbumin), a tetrameric protein synthesized by the liver that normally transports vitamin A and thyroid hormone. ATTR amyloidosis can be further divided into two subtypes:
Wild-type amyloidosis (wtATTR amyloidosis) – Wild-type transthyretin amyloidosis (previously known as senile systemic amyloidosis) is caused by the deposition of misfolded wild-type (normal) transthyretin.
Hereditary amyloidosis (hATTR amyloidosis) – Hereditary transthyretin amyloidosis is caused by gene mutations in the transthyretin gene (TTR) that lead to abnormal transthyretin formation. The typical transmission of hATTR is autosomal dominant inheritance with variable penetrance, and there are more than 120 known mutations of TTR associated with hATTR amyloidosis.
Light chain amyloidosis (AL amyloidosis) – Light chain amyloidosis (AL amyloidosis; also known as primary systemic amyloidosis) results from deposition of misfolded immunoglobulin light chains from a plasma cell dyscrasia.
Other types of amyloid – Rare causes of cardiac amyloidosis include serum amyloid A amyloidosis, hereditary apolipoprotein A-1, and apolipoprotein A-4 amyloidosis.
Clinical manifestations – The clinic phenotype varies, and an often-generic clinic presentation makes it difficult to establish a diagnosis.
Presentations at age ≥60 years are most common. Each transthyretin mutation is associated with its own age range (from 30 to 70 years) and the particular risk for cardiomyopathy varies. The main manifestations of ATTR amyloidosis are cardiac.
Electrocardiogram – Discordance between increased left ventricular wall thickness (on cardiac imaging ie echocardiography) and QRS voltage, which is often reduced, is the classic sign of cardiac amyloidosis. However, this has low sensitivity, and the prevalence of low voltage shows significant variation, depending on the cause, with less frequency in patients with ATTR amyloidosis (20 percent) than in patients with AL amyloidosis (60 percent).
When to suspect cardiac amyloidosis – Cardiac amyloidosis should be suspected in patients with:
Unexplained LV hypertrophy (with or without heart failure [HF]).
Aortic stenosis with features associated with cardiac amyloidosis (such as presence of low-flow, low-gradient aortic stenosis and/or echocardiographic detection of impaired longitudinal strain [eg, mitral annular S’ ≤6 m/sec]).
Symptoms or signs typical of AL or ATTR amyloidosis and HF.
A condition highly associated with cardiac amyloidosis (eg, systemic AL amyloidosis, ATTR-related peripheral neuropathy, or ATTR mutation carrier state).
Diagnosis – For patients with any of the above features, cardiovascular magnetic resonance (CMR) imaging is recommended.
If CMR findings suggest cardiac amyloidosis, testing for evidence of monoclonal protein using serum protein immunofixation, urine protein immunofixation, and serum free light chain ratio analysis is the next step.
If monoclonal protein is identified, hematology referral, tissue biopsy and bone marrow biopsy are indicated.
If monoclonal protein is not identified, further evaluation is based upon the results of bone tracer cardiac scintigraphy.
If CMR is not suggestive of cardiac amyloidosis, cardiac amyloidosis is unlikely and other causes of LVH and/or HF should be considered.
Treatment of specific complications of cardiac amyloidosis
Atrial fibrillation
Rate and rhythm control – In patients with atrial fibrillation, rate control should be prioritized over rhythm control. For most patients, amiodarone is recommended for rate control. In patients in whom amiodarone is not an option for therapy, low-dose digoxin or low-dose beta blockers are alternatives.
Anticoagulation – In patients with cardiac amyloidosis and atrial fibrillation or atrial flutter, anticoagulation is recommended. Standard risk estimators (eg, CHA2DS2VASC) are not validated in amyloidosis.
Cardioversion – In patients with AL or ATTR cardiac amyloidosis who require cardioversion for symptomatic management, transesophageal echocardiography (TEE) is recommended prior to cardioversion rather than no transesophageal echocardiography prior to cardioversion. This is to exclude the presence of emboli.
Conduction system disease – In patients with cardiac amyloidosis, general indications for cardiac pacing are recommended.
Specific treatment for ATTR amyloidosis
Medical therapy – In patients with ATTRwt or ATTRv (where “v” indicates “variant”) cardiac amyloidosis and New York Heart Association (NYHA) functional class I to III HF symptoms, tafamidis is recommended rather than no disease specific therapy, and tafamidis is preferrable to diflunisal. Diflunisal is poorly tolerated and has unclear efficacy in patients with cardiac amyloidosis.
Therapy for heart failure – In patients with ATTR cardiac amyloidosis with either HFrEF (HF with reduced ejection fraction) or HFpEF (HF with preserved ejection fraction), there are no specific recommendations for HF therapy other than general HF treatment measures and diuretics for volume overload.
In patients with refractory HF, therapeutic options include palliative care, heart transplantation, mechanical circulatory support, and continuous inotrope infusion.
Liver transplantation – This only has proven benefit for patients with familial amyloid polyneuropathy. Potential benefit for cardiac amyloidosis is controversial.
Prognostic indicators
The first published staging system for patients with ATTRwt cardiac amyloidosis is based on serum levels of NT-proBNP and cardiac troponin T.
The second staging system, validated in patients with ATTRwt or ATTRv, is based on serum levels of NT-proBNP and eGFR.
Adapted from the following UpToDate articles:
Cardiac amyloidosis: Epidemiology, clinical manifestations, and diagnosis by Marianna Fontana, MD
Cardiac amyloidosis: Treatment and prognosis by Marianna Fontana, MD
By Dr. Shifa Younus Nishtar Medical College, Multan, Pakistan. Amin H. Karim MD
Case Presentation:
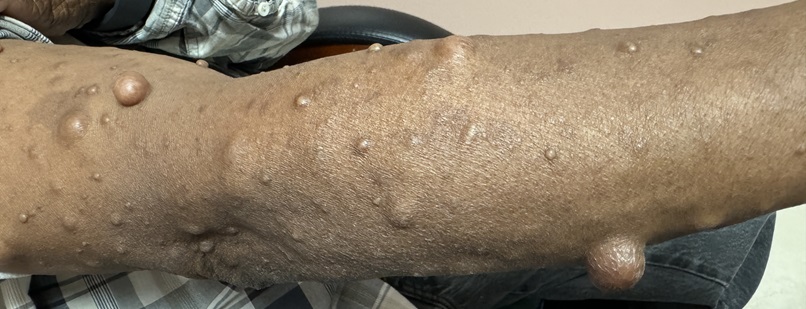
A 65-year-old patient, presents in the office with multiple raised lesions all over his body more so on face and extremities. The patient has had these lesions since childhood. He had a few lesions in the beginning, but these progressively increased in number with time. The Patient complains of high blood pressure readings for one year. There is no history of neurological tumors, intellectual disability, or renal involvement.
Family History: The Patient has a family history of similar lesions in his son and grandson. Both have these lesions with associated symptoms.
Physical Examination: Extremity Examination: There are multiple lesions which are raised, mobile, and painless with no associated ulcers or change in coloration of the surrounding skin.
Diagnosis: Based on the symptoms and history, Mr. Mccray was diagnosed with Neurofibromatosis type 1 or Von Recklinghausen’ disease.
Neurofibromatosis type 1 or Von Recklinghausen’s Disease:
Neurofibromatosis type 1 (NF-1) was formerly known as Von Recklinghausen’s disease after the German pathologist, Friedrich Daniel von Recklinghausen (1822-1910). He was the student of a famous pathologist Rudolph Virchow in University of Berlin. and served as the professor of pathology in various universities. He is best known for his description of three disorders: multiple neurofibromatosis, osteitis fibrosa cystica, and hemochromatosis.[1]
NF-1 is one of the most common neurocutaneous genetic disorders with autosomal pattern of inheritance. It accounts for 1 out of 3000 live births. It is caused by the mutations in NF-1 gene, present on chromosome 17, which encodes a cytoplasmic protein, neurofibromin present in neurons, leukocytes, oligodendrocytes, and Schwann cells. [2]
NF-1 is an age specific disease which becomes apparent during infancy, but it can also appear in adulthood due to hormonal changes. The severity of the disease varies from person to person. However, about 60% of the patients present with mild disease with no effect on their daily life. Neurofibromas, the most common presentation related to NF-1, are the peripheral nerve sheath tumors arising from Schwann cells and fibroblasts. [3]
NF-1 comprises of multiple manifestations ranging from cutaneous lesions to serious neurological and systemic manifestations which include:
Craniofacial manifestations: Orbital dysplasia Intraosseous neurofibroma of maxilla and mandible Pheochromocytoma. Attention deficit hyperkinetic disorder.
NF-1 can lead to some serious complications including bone tumors and peripheral nerve damage that may also lead to vision loss. Some neurofibroma may rarely degenerate into malignant tumors. However, early diagnosis and treatment plays an important role in improving the prognosis. The National Institute of Health Consensus Development Conference in 1988 put forth a diagnostic criterion for NF-1. If the patient has two or more of the following:
Six or more café au lait macules
Two or more neurofibromas of any type or one plexiform neurofibroma
Freckling in the axillary or inguinal regions
Optic glioma
Two or more iris Lisch nodules
Distinctive osseous lesion such as sphenoid dysplasia
Family history of the first-degree relative with neurofibromatosis. [4]
Other diagnostic techniques include:
Slit-lamp examination may be performed on children to look for Lisch nodules.
T2 weighted MRI of the brain and spine which may reveal hyperintense lesions due to aberrant myelination or gliosis of neurons. [5]
Biopsy of the neurofibromas which may reveal non-encapsulated tumor composed of fascicles of slender, spindle-shaped cells.
Prenatal diagnosis of NF-1 can be made by genetic testing.
The management of neurofibromatosis is done by a multidisciplinary team which includes dermatologists, neurologists, oncologists, pediatricians, and genetic counselors. Café au lait spots and cutaneous neurofibromas are particularly benign and do not require treatment. However, symptomatic lesions or ones with malignant potential may require surgical excision but their recurrence is very common. [6] Apart from the mainstay treatment by surgery there are certain medical treatments that target at the molecular level and inhibit the pathway involved in pathophysiology of neurofibromas. These include selumetinib, mirdametinib, binimetinib which have proven to be helpful in decreasing the overall volume of neurofibromas and providing symptomatic relief. NF-1 has a considerable impact on general quality of life (QoL) of the patients due to it dermatological manifestations. Therefore, psychosocial support is of immense importance in such patients and cannot be neglected. [7]
2.Ghalayani P, Saberi Z, Sardari F. Neurofibromatosis type I (von Recklinghausen’s disease): A family case report and literature review. Dent Res J (Isfahan). 2012;9(4):483-488.
3.Nallanchakrava S, Mallela MK, Jeenepalli VSK, Niharika HM. A rare case report of neurofibromatosis type 1 in a 12-year-old child: A 15-month follow-up. J Oral Maxillofac Pathol. 2020;24(Suppl 1):S106-S109. doi:10.4103/jomfp.JOMFP_35_20
4. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906
5. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906
6. Taylor LA, Lewis VL Jr. Neurofibromatosis Type 1: Review of Cutaneous and Subcutaneous Tumor Treatment on Quality of Life. Plast Reconstr Surg Glob Open. 2019;7(1):e1982. Published 2019 Jan 18. doi:10.1097/GOX.0000000000001982
7. Taylor LA, Lewis VL Jr. Neurofibromatosis Type 1: Review of Cutaneous and Subcutaneous Tumor Treatment on Quality of Life. Plast Reconstr Surg Glob Open. 2019;7(1):e1982. Published 2019 Jan 18.
By Farhat Soluade Second Year P.A. Student, West Coast University. Amin H. Karim MD
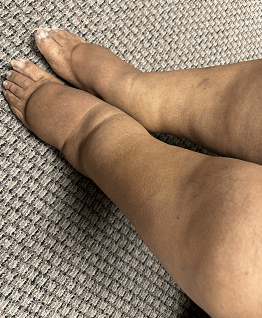
Case Presentation: A 70 year old female presents with a chief complaint of swelling and discomfort in both lower extremities. She reports that the swelling has been progressively worsening over the past six months, prompting her to seek medical attention. The swelling is associated with a feeling of heaviness and tightness in her legs. Approximately six months ago, Mrs. Smith noticed swelling in both her legs, which has been gradually increasing. The swelling is more pronounced in the evening and is not relieved by elevation. She denies any recent trauma, infection, or prolonged immobilization.
Cardiovascular and Pulmonary History: The patient has a history of hypertension for the past 5 years, which is well-controlled with antihypertensive medications. She denies any chest pain, shortness of breath, or palpitations. Pulmonary history is unremarkable.
Family History: There is no significant family history of cardiovascular diseases, deep vein thrombosis, or lymphatic disorders.
Physical Examination: Vital Signs: Blood pressure: 130/80 mmHg;Heart rate: 72 bpm; Respiratory rate: 16 breaths/min; Temperature: 98.6°F (37°C) Lower Extremities: Lymphostatic elephantiasis; Bilateral non pitting edema extending up to the knees : Skin appears normal with no signs of cellulitis or ulceration; Tenderness on palpation.
Cardiovascular and Respiratory Examination: No murmurs, gallops, or respiratory distress Clear lung fields bilaterally
Doppler Ultrasound of Lower Extremities: Rule out deep vein thrombosis (negative findings) Lymphoscintigraphy: Confirm the diagnosis of lymphedema by demonstrating impaired lymphatic drainage in the lower extremities
Diagnosis: Stage III Lower extremity lymphedema, possibly secondary to age-related changes or an underlying, yet-to-be-determined cause.
LYMPHEDEMA
The abnormal buildup of interstitial fluid and fibro adipose tissues brought on by congenital infection, congenital abnormalities, or injury of the lymphatic system is known as lymphedema. Although it can occur anywhere on the body, lymphedema most commonly affects the extremities. A variety of hereditary syndromes and disorders can cause a defect in the conduits that carry lymph, which leads to primary lymphedema. Acquired disorders can cause a defect or obstruction in lymph-carrying conduits, which can lead to secondary lymphedema.
The patient’s medical history may disclose factors that contributed to the development of lymphedema. Primary causes include genetic syndromes such as Milroy disease caused by vascular endothelial growth factor receptor (VEGFR-3 mutation), lymphedema-distichiasis syndrome (FOXC2 mutation), and hypotrichosis-lymphedema-telangiectasia syndrome (SOX 18 mutation). Primary causes may also include functional causes such as valvular insufficiency or paralysis. The most common secondary cause includes filariasis which is a zoonotic infection caused by parasitic nematodes (due to Wuchereria bancrofti). Malignancy and malignancy-related treatments such as neoplastic destruction of the lymph vessels or surgical removal of lymph nodes are the second most common secondary cause. Burns, recurrent bacterial infections, and wounds to extremities can cause disruption or damage to lymphatic channels. Chronic venous insufficiency and occlusive peripheral arterial disease are also common causes of lymphedema.
The thoracic duct is the main conduit that connects the lymphatic vasculature, a low-pressure system, to the central circulation. The larger conduits in the system contain contractile smooth muscle. The lymphatic vasculature connects interstitial spaces with lymphoid organs like the spleen and lymph nodes. Lymphedema is the characteristic swelling (usually in the extremities) caused by the accumulation of excess water, plasma proteins, parenchymal/stromal cell products, and extravascular blood cells in the extracellular space when there is an imbalance between the rate of lymph production and its removal through the lymphatic vascular channels. Lymphedema happens because of a low-output failure that results in a reduction in lymphatic transport. Progressive fibrosis, cutaneous thickening, hypercellularity, and a pathologic rise in subcutaneous and subfascial adipose tissue can all result from lymphedema over time.
Diagnosis is normally made by clinical history and physical examination. Patients may be asymptomatic or symptomatic depending on the stage with symptoms of impaired mobility of the affected limb or discomfort. Patients with lower extremity lymphedema initially present with unilateral swelling in the dorsal aspect of the foot and after a year it may progress to the proximal aspect of the foot. The edema may spread circumferentially over time. Later on in the disease, nonpitting edema will occur following fibrotic changes in subcutaneous tissue. The skin may be hyperpigmented with increased skin turgor and hyperkeratotic. Physical examination of the skin and extremities will reveal edema which is usually pitting in the early stages but may become non pitting in later stages and be linked to fibrotic changes and the build-up of adipose tissue. The skin should be checked for any complications such as cellulitis. A complete metabolic profile may be ordered to rule out alternative causes of edema such as renal failure. Lymphoscintigraphy may be indicated for cases where the diagnosis is in question. Differential Diagnosis includes edema from a systemic origin such as heart failure, deep vein thrombosis, medication-induced edema, or obesity.
International Society of Lymphology (ISL) staging of lymphedema:
Stage 0 or Ia: Asymptomatic.
Stage I: Edema is present and limb elevation can reduce the edema.
Stage II: Swelling, pitting edema, progresses to nonpitting edema. Elevation does not reduce swelling.
Complex decongestive physiotherapy (CDPT) also known as Combined Physical Therapy (CPT) should be offered for the management of lymphedema. The goal of CDPT is to reduce limb volume and to support the skin’s health. There are 2 phases to CDPT. Phase 1 involves manual lymphatic drainage, compression, multilayered bandage wrapping, and range-of-motion exercises. Phase 2 includes using fitted clothes to stop the fluid from re-acclimating, remedial exercise, and as many light messages as necessary. Diuretics are normally not indicated for treatment unless there are specific comorbidities that require diuretic therapy. If obesity is a factor, exercise and weight management should be implemented. Surgery can be considered if patients are unresponsive to other therapies and if lymphedema impairs quality of life and function. Unfortunately, despite conservative measures, lower extremity lymphedema frequently worsens. Due to the time commitment, ongoing treatment requirements for the rest of one’s life, the scarcity of certified lymphedema therapists, the cost, insurance coverage concerns, and the discomfort caused by wearing bulky compression garments in hot weather, patient compliance can be challenging.
Author: Farhat Soluade
References:
Van Zanten M, Mansour S, Ostergaard P, et al. Milroy Disease. 2006 Apr 27 [Updated 2021 Feb 18]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1239/
A 72 year old dentist presented with several months of increasing shortness of breath. Echocrdiogram is as follows:
Severe left ventricular hypertrophy
Echocardiogram showed global ejection fraction of 50%; Grade III diastolic dysfunction with restrictive LV filling pressures; PA systolic pressure of 65-70 mm Hg. a specled appearance of the myocardium consistent with cardiac amyloidosis.
Coronary angiogram did not reveal any occlusive coronary disease.
Right ventricular endomyocardial biopsy showed a variable deposiiton of acellular material within the interstitium and replacing some myocardial fibres. Congo red stain and fluorescence microscopy with the Texas Red filter demonstrated strong positivity further supportive of amyloid deposition. The paraffin block was sent to Mayo Clinic for amyloid subtyping. it showed findings most consistent with age related amydoidosis since it did not detect amino acid sequence abnormality in the transthyretin protein.
November 2023: Four years after the above diagnosis patient is stable on treatment. He has a implantable automatic defibrillator and the current global ejection fraction is in the 45-50% range.
North Pole is a small Alaskan city, near Fairbanks. It’s known for its year-round Christmas decorations, including candy cane–striped street lights. Santa Claus House is a Christmas store with walls covered in children’s letters to Santa and a huge Santa statue outside. Streets have names like Kris Kringle Drive and Mistletoe Lane;
It has a population of about 2300 people who are kind enough to volunteer for parents to send them gifts for children and they in turn mail it back to them via their post office so that they are stamped by the name “North Pole Post Office”.
Now, what has this to do with a Cardiology forum. I will tell you a little story with some history to it.
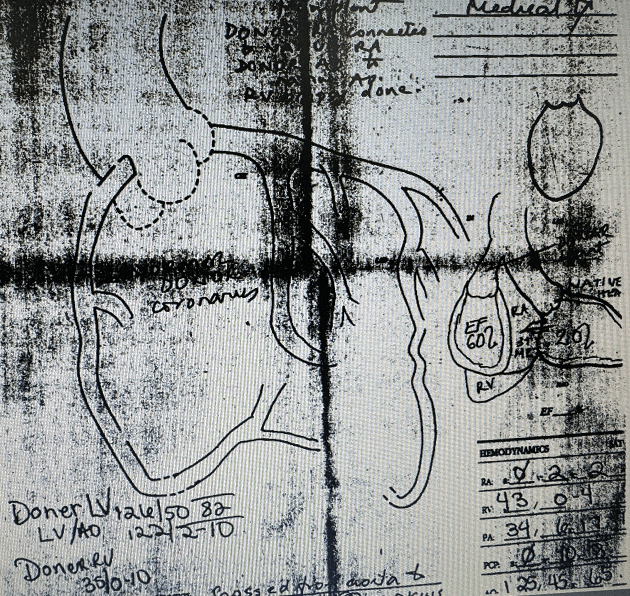
In late 1990s a 65 year old male patient from North Pole Alaska came to me with a complaint of shortness of breath. We found that he had end stage low EF congestive cardiac failure and after the usual workup enlisted him for heart transplant. He was lucky to get one and Dr. Howard Frazier ( who to this day has done the most heart transplants on the Planet) performed a rather atypical heart transplant on him. Instead of removing the entire front part of the recipient heart and suturing the donor heart over it, he did what was called ” a piggy back transplant” in which he left the native heart alone and connected the donor heart to the aorta and the RA/RV , thus creating a sort of ” Left ventricular Assist Device”. I am not sure what the exact reason for this experiment was but apparently he knew what he was doing The patient did well and went home and continued to improve for several months off this piggy back heart. He came back from Alaska and I did a cardiac cath on him as was the protocol along with an endomyocardial biopsy of the donor heart to check for rejection. I got the most strange experience of looking for two sets of coronaries and two left ventriculograms in the same person. The native coronaries were diseased but the donor coronaries were normal angiographically.
The patient continued to do well and became quite functional and tolerated his anti rejection regimen. One day, in 1999, he noticed a growth on his year and consulted his primary care doctor in Alaska who told him not to worry about it. Tragically, it turned out to be a melanoma that quickly spread to his brain and proved fatal. No wonder, to this day I still have his extended family and friends come to Houston for treatment as they do not trust the doctors there.